
Gute Laborpraxis (Good Laboratory Practice, GLP) entscheidet in MedTech-Projekten oft früher über Erfolg oder Rückschlag, als viele Teams erwarten. Nicht beim Schreiben des Abschlussberichts (Final Report), sondern in dem Moment, in dem die erste Messung dokumentiert wird.
Regulatoren prüfen nicht nur das Ergebnis einer präklinischen Studie, sondern vor allem, ob Fragestellung, Durchführung, Datenbasis und Dokumentation belastbar und für den jeweiligen regulatorischen Kontext verwertbar sind. Die Kernfrage im Audit lautet selten „Haben Sie GLP angewendet?“. Sie lautet: „Können Sie belegen, was wann warum getan wurde und wer dafür verantwortlich war?“
Dieser Artikel zeigt, welche 10 GLP practices-Kernelemente Auditoren tatsächlich prüfen, wo in der Praxis die größten Lücken entstehen, wann ein strukturierter GLP-like-Ansatz ausreicht und wann nicht. Die Grundsätze der guten Laborpraxis (GLP) bilden dabei den regulatorischen Referenzrahmen.
Hinweis zur Artikelreihe: Dieser Artikel beantwortet die operative Frage, was genau umzusetzen ist, wenn ein Team sich für einen GLP-like-Ansatz entschieden hat. Die vorgelagerte strategische Frage, ob GLP oder GLP-like die richtige Wahl ist, wann GLP regulatorisch zwingend wird und was beide Ansätze kosten, behandelt der Artikel GLP vs. „GLP-like“ in MedTech-Studien. Beide Artikel ergänzen sich: Strategie dort, operative Umsetzung hier.
GLP practices: Was Behörden an präklinischen Reports wirklich prüfen
Regulatorisch lautet die eigentliche Frage nicht: „War das Ergebnis positiv?“ Sie lautet: Kann diese Evidenz belastbar für eine regulatorische Entscheidung herangezogen werden?
Behörden wie die FDA oder Benannte Stellen nach EU MDR (Medical Device Regulation) bewerten die Entstehungslogik eines Berichts. In der Praxis arbeiten Auditoren entlang einer Evidenzlogik mit vier Blöcken:
- War die Studie vorab sinnvoll definiert?
- Sind Durchführung und Datenfluss beherrscht?
- Wurden Abweichungen transparent bewertet?
- Lässt sich die Studie später rekonstruieren?
Wer diese vier Blöcke sauber aufstellt, hat die härtesten Fragen oft bereits beantwortet. Für den Medizintechnik-Sektor (MedTech) ist das besonders relevant: Präklinische Daten stützen interne Entscheidungen, fließen in technische Dokumentationen ein und werden oft gleichzeitig regulatorisch hinterfragt.
Grundsatz: Im Kern sind GLP good laboratory practice und GLP-like-Prinzipien Vertrauensmechanismen. Sie sorgen dafür, dass aus Messwerten und Beobachtungen eine belastbare Evidenzkette wird, beginnend mit der ersten dokumentierten Handlung.
Das häufigste Missverständnis: Gute Ergebnisse reichen aus
Viele Projektteams konzentrieren sich auf die Qualität der experimentellen Daten. Auditoren beginnen aber auf der Systemebene: Rollen, Prozesse, Dokumente. Ein technisch sauberes Experiment mit lückenhafter Dokumentation scheitert im Review genauso wie ein schlecht geplantes Studiendesign.
Ein starker Abschlussbericht kann fehlende Rohdatenlogik, unklare Verantwortlichkeiten oder nicht bewertete Abweichungen nicht heilen. Er kann Lücken sprachlich glätten, aber nicht regulatorisch schließen. Audit-Fähigkeit beginnt bei der Planung und Konzeption, nicht im Verfassen des Berichts.
Das zweite Missverständnis betrifft GLP-like. Der Begriff klingt nach bewusst informellem Zustand. Praktisch funktioniert er aber nur dann, wenn die relevanten Kontrollpunkte verbindlich sind: sauberer Studienplan, definierte Rohdaten, belastbare SOPs (Standardarbeitsanweisungen), dokumentierte Qualifikation der Beteiligten, unabhängige Qualitätssicherung (QA), kontrollierte Systeme, funktionierender Archivpfad.
GLP-like im MedTech-Kontext: kein Kompromiss, sondern ein klarer Rahmen
GLP-like ist kein offizieller Rechtsbegriff. Aber er beschreibt eine regulatorische Realität: Nicht jede präklinische Studie im MedTech-Umfeld erfordert in der Regel einen formell überwachten GLP-Rahmen mit unabhängiger Qualitätssicherung sowie Inspektionen bzw. Compliance-Monitoring der Prüfeinrichtung nach national umgesetzten OECD-GLP-Grundsätzen (Organisation for Economic Co-operation and Development).
Was Behörden dennoch erwarten, ist Substanz. Eine GLP-like-Studie muss die relevanten Kernelemente der GLP practices abbilden, auch wenn sie nicht formell unter das GLP-Regime fällt. GLP-Konformität (GLP compliance) ist dabei kein einmaliges Projekt, sondern ein kontinuierlicher Nachweis, dass Prozesse, Daten und Dokumentation den regulatorischen Erwartungen entsprechen. Wer diese Grenze kennt und die richtigen Strukturen früh aufbaut, vermeidet nachträgliche Nachbesserungen und unnötige Folgestudien.
Die 10 GLP-like Kernelemente für audit-fähige Reports
Ein praktikabler Ansatz für GLP practices steht und fällt mit zehn Kernelementen. Sie übersetzen die regulatorischen Erwartungen in operative Prüfpunkte, die auch ohne vollständige GLP-Zertifizierung umsetzbar sind. Das gemeinsame Ziel: ein prüfungsfähiger Abschlussbericht (audit-ready report), der einer behördlichen Prüfung standhält, unabhängig davon, ob eine formelle GLP-Zertifizierung vorliegt oder nicht. Jedes der folgenden Kernelemente trägt direkt zur Verteidigungsfähigkeit der Studie bei.
| 01 |
Klare Fragestellung, Endpunkte & FreigabekriterienVor Studienstart muss schriftlich definiert sein, welche Frage beantwortet werden soll. Vorab festgelegte Endpunkte und Freigabekriterien bilden die Grundlage jeder verteidigungsfähigen Evidenz und sind die erste Kontrollfrage jedes Auditors. |
| 02 |
Studienplan mit VersionsmanagementJede GLP-like-Studie beginnt mit einem schriftlichen Studienplan, der vor Studienbeginn erstellt wurde, nicht danach. Der Plan muss versioniert, datiert und abgezeichnet sein. Protokolländerungen sind nur nummeriert und mit der Bewertung der regulatorischen Auswirkung zulässig. Was nicht im Plan steht, ist im Audit kaum zu verteidigen. |
| 03 |
Rohdaten-Definition nach ALCOA+Rohdaten sind alle Originaldaten, aus denen Ergebnisse abgeleitet werden, bspw. Messwerte, Bilder, Kalibrierprotokolle, handschriftliche Notizen. Sie müssen nach den ALCOA+-Kriterien erstellt sein: A – attributierbar (zuordenbar), L – leserlich, C – zeitnah erfasst, O – original, A – korrekt, sowie vollständig, konsistent, dauerhaft und verfügbar. Überschriebene Excel-Dateien gehören zu den häufigsten schwerwiegenden Befunden – definierte Rohdatenpfade sind daher ein unverzichtbarer Bestandteil. |
| 04 |
Prüfgegenstand-Charakterisierung & lückenlose Proben- und MaterialketteAuditoren und Benannte Stellen prüfen, ob die getesteten Muster repräsentativ für das spätere Marktprodukt sind. Der Bericht muss Materialzusammensetzung, Chargenrückverfolgung und Herstellungshistorie inkl. Reinigung und Sterilisation lückenlos dokumentieren. Fehlt diese Dokumentation, kann die regulatorische Verwertbarkeit der Studie deutlich eingeschränkt sein. |
| 05 |
SOPs, Gerätekalibrierung & SystemvalidierungSOPs (Standardarbeitsanweisungen) für studienkritische Prozesse müssen gelebte Praxis abbilden, nicht nur existieren. Alle Messinstrumente werden regelmäßig kalibriert und in Geräte-Logbüchern dokumentiert. Computergestützte Systeme erfordern zusätzlich eine Computervalidierung (CSV) inklusive Softwareversion und Prüfspuren. Viele GLP practices-Konzepte berücksichtigen diesen Punkt erst spät. |
| 06 |
Unabhängige Qualitätssicherung (QA)Die Qualitätssicherung prüft Studienplan, Protokolle und Abschlussbericht unabhängig vom Studienbetrieb. Im GLP-like-Rahmen kann diese Funktion extern beauftragt werden. Entscheidend ist die Nachweisbarkeit: Wer hat was geprüft, wann und mit welchem Ergebnis? Ohne dokumentierten Nachweis der Prüfungsschritte durch die Qualitätssicherung ist die Audit-Fähigkeit des Reports regelmäßig deutlich geschwächt. |
| 07 |
Studienleiter, Rollen & TrainingsnachweisJede Studie braucht einen namentlich benannten Studienleiter (Study Director), der Studienplan und Abschlussbericht abzeichnet. Alle Beteiligten belegen Ausbildung, Training und Eignung für ihre konkrete Aufgabe mit Datum, SOP-Inhalt und Unterschrift. Wer war beteiligt und war diese Person dafür qualifiziert? Diese Frage gehört in Audits häufig zu den frühen Prüfpunkten. |
| 08 |
Vollständige Abweichungs-Dokumentation & CAPA-Prozess (Korrektur- und Vorbeugungsmaßnahmen)Abweichungen vom Studienplan passieren in jeder Studie. Auditoren erwarten zeitnahe Erfassung, eine Bewertung der regulatorischen Auswirkung und eine klare Entscheidung: akzeptiert oder eskaliert. Datenrelevante Abweichungen müssen dokumentiert und bewertet werden; bei kritischen, wiederkehrenden oder systemischen Ursachen sollte eine CAPA mit Termin und Wirksamkeitsnachweis folgen. Offene CAPAs bei der regulatorischen Einreichung signalisieren systemische Dokumentationsprobleme und sind eine Einladung zur vertieften Prüfung. |
| 09 |
Statistische Validität & nachvollziehbare DatenauswertungAuswertungsmethoden müssen fachlich geeignet, vorab geplant und transparent dokumentiert sein. Das gilt auch für den Umgang mit statistischen Ausreißern, Wiederholungen und negativen Ergebnissen. Wer statistische Verfahren erst nach der Datenerhebung wählt, riskiert den Vorwurf des Post-hoc-Designs, was ein klassisches Signal für vertiefte Prüfung durch Regulatoren darstellt. |
| 10 |
Abschlussbericht, Stellungnahme der Qualitätssicherung, Archivierung & BehördeninspektionsbereitschaftDer Abschlussbericht referenziert alle Primärdaten, fasst Abweichungen zusammen und dokumentiert den CAPA-Status. Der Abschlussbericht wird vom Studienleiter freigegeben. Im formellen GLP-Rahmen gehört eine dokumentierte QA-Stellungnahme zwingend in den Abschlussbericht. Im GLP-like-Kontext sollte zumindest eine nachvollziehbar dokumentierte, unabhängige Qualitätsprüfung Bestandteil des Reports sein. Anschließend folgt die zugangsbeschränkte Archivierung aller relevanten Studienunterlagen. Die konkreten Aufbewahrungsfristen sollten jeweils nach anwendbarem Regime, Einreichungskontext und Produkttyp festgelegt werden. Auditbereitschaft endet nicht mit der Unterschrift, sondern Dokumente müssen jederzeit zügig abrufbar sein. |
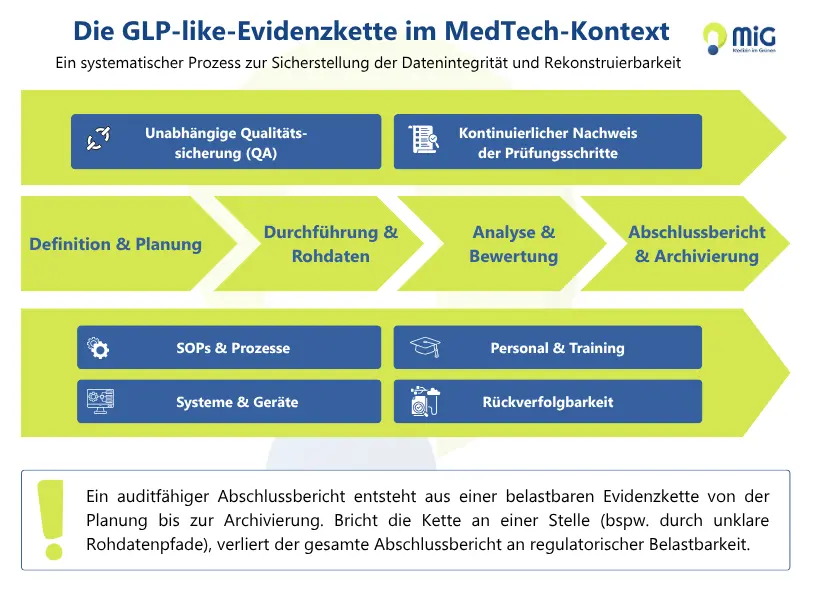
Grafik: „GLP practices Evidenzkette im MedTech-Kontext“
Wo in der Praxis die größten Lücken entstehen
In frühen MedTech-Programmen liegen die größten Schwächen selten in spektakulären Fehlern. Es sind kleine Brüche in der Evidenzkette, die später große Konsequenzen haben.
Unscharfer Rohdatenstatus
Versuchsdaten liegen vor, sind aber nicht eindeutig als Primärdaten markiert und nicht sauber mit der Auswertung verknüpft. Auditoren können dann nicht nachvollziehen, welcher Datensatz tatsächlich die Berechnungsgrundlage war. Dieses Problem gehört in der Praxis zu den häufigsten und lässt sich mit klaren Rohdatenregeln meist früh entschärfen.
Schwaches Änderungs- und Abweichungsmanagement
Änderungen wurden vorgenommen, aber nicht so dokumentiert, dass später noch klar ist, warum sie nötig waren und welche Auswirkung sie hatten. Unabhängig von der experimentellen Qualität der Studie gehört eine fehlende oder lückenhafte Dokumentation der Abweichungen zu den häufigsten schwerwiegenden Befunden in präklinischen Audits.
Informelle Qualitätssicherung ohne Nachweis der Prüfungsschritte
Die Prüfung hat stattgefunden, aber kein dokumentierter Nachweis der Prüfungsschritte durch die Qualitätssicherung belegt sie. Ohne Nachweis der Unabhängigkeit des Prüfergebnisses zählt die Prüfung durch die Qualitätssicherung im Audit nicht. Hinzu kommen fehlende Prüfspuren bei computergestützten Systemen und schwach validierte Software als unsichtbare Stolpersteine, welche die GLP practices-Evidenzkette erheblich schwächen.
Wer diese drei Punkte früh absichert, verhindert, dass kleine Lücken im Report oder Audit zu strukturellen Problemen werden.
Ihr Selbstcheck: die GLP-like Bewertungsmatrix
Die 10 Kernelemente lassen sich direkt als Selbstcheck im Projektteam einsetzen. Bewerten Sie jeden Punkt mit einer Ampellogik:
- Grün = vollständig vorhanden und dokumentiert
- Gelb = vorhanden, aber lückenhaft oder nicht aktuell
- Rot = fehlt vollständig
Drei oder mehr rote Punkte sind ein starkes Indiz dafür, dass die Verteidigungsfähigkeit des Reports in einer Prüfung, unabhängig von der experimentellen Qualität, deutlich sinkt. Besonders kritisch ist dabei der Status der Rohdaten (raw data): Lassen sich alle Primärdaten lückenlos auf ihre Herkunft zurückverfolgen und sind sie eindeutig mit der Auswertung verknüpft? Je früher diese Lücken identifiziert werden, desto geringer der Aufwand zur Nachbesserung.
Wann der Wechsel zu vollständigem GLP sinnvoll wird
GLP-like ist kein Dauerzustand. Es gibt Konstellationen, in denen der Wechsel zu einem vollständigen GLP-Standard nach OECD-Prinzipien notwendig oder strategisch richtig ist:
- FDA-regulatorische Einreichung mit IDE (Investigational Device Exemption)- oder PMA (Premarket Approval)-Anforderungen
- MDR-Einreichungen für Hochrisikoprodukte, bei denen sicherheitsrelevante tierexperimentelle Daten eine tragende Rolle für die regulatorische Argumentation spielen
- Wiederholt auftretende Beanstandungen in GLP-like-Reports
- Studienpartner oder Auftragsforschungsorganisation (CRO) mit GLP-Zertifizierung geplant
Wenn Daten nicht mehr nur intern genutzt, sondern eine regulatorische Entscheidung direkt stützen sollen, steigen die Erwartungen in GLP-Inspektionen und externen Audits deutlich. Dann reicht ein pragmatischer GLP-like-Stand oft nicht mehr aus.
Der entscheidende Zeitpunkt für diese Entscheidung liegt nicht nach dem Studiendesign, sondern davor. Eine nachträgliche Anhebung eines GLP-like-Datensatzes auf GLP-Niveau ist in aller Regel nicht möglich. Frühe Projektabgrenzung mit einem erfahrenen Berater für regulatorische Angelegenheiten spart hier erheblich Zeit und Budget.
Fazit
Viele MedTech-Teams diskutieren zu früh über das Label GLP oder non-GLP und zu spät über die eigentliche Frage: Ist unsere Evidenzlogik belastbar?
Genau deshalb sind GLP practices so wertvoll. Wer Fragestellung, Plan, Rollen, SOPs, Training, Datenintegrität, Testartikel, Geräte, Statistik und Archivierung konsequent absichert, schafft einen GLP-like-Reifegrad, der frühe Entwicklung mit späterer Audit-Fähigkeit (audit readiness) verbindet und regulatorische Überraschungen vermeidet.
GLP-like Quick Check
Wie viele der 10 Kernelemente sind in Ihrem Report wirklich vollständig?
Im GLP-like Quick Check analysieren wir gemeinsam Ihren aktuellen Status und identifizieren konkrete Lücken mit Blick auf Nachvollziehbarkeit, Datenintegrität und regulatorische Anschlussfähigkeit.
Häufig gestellte Fragen zu GLP practices
Was ist der Unterschied zwischen GLP und GLP-like?
GLP (Gute Laborpraxis) ist ein formell regulierter Standard nach OECD-Grundsätzen mit behördlicher Anerkennung, Pflicht zur unabhängigen Qualitätssicherung und GLP-Inspektion der Prüfeinrichtung. GLP-like beschreibt einen pragmatischen Ansatz, der die wesentlichen Strukturelemente der GLP practices umsetzt, ohne unter das formelle GLP-Regime zu fallen. Der wesentliche Unterschied liegt im Umfang der Verpflichtungen: Vollständiges GLP setzt behördlich überwachte GLP-konforme Prüfeinrichtungen, unabhängige Qualitätssicherung und GLP-Inspektionen bzw. Compliance-Monitoring voraus. GLP-like hingegen setzt die inhaltlichen Kernelemente um, bleibt aber unterhalb dieser formellen Schwelle. GLP-like ist kein anerkannter Rechtsbegriff, aber eine in der Praxis gängige Kategorisierung.
Reicht GLP-like für die Einreichung relevanter Daten aus?
Für frühe explorative Arbeiten häufig ja. Für zentrale sicherheitsrelevante Daten reicht ein GLP-like-Ansatz dauerhaft oft nicht aus. Das hängt von Geräteklasse, Risikoniveau und Studientyp ab. Bei Unsicherheit empfiehlt sich eine frühzeitige Klärung mit einem Berater für regulatorische Angelegenheiten oder der Benannten Stelle, bevor das Studiendesign fixiert wird.
Was ist der häufigste blinde Fleck bei GLP practices?
Meistens sind es keine sichtbaren Dokumente, die fehlen, sondern unklare Rohdatenpfade, informelle Qualitätssicherung oder schwach kontrollierte Softwaresysteme. Fehlende Prüfspuren und unklare Rollenrechte schwächen die Evidenzkette erheblich, ohne dass das auf den ersten Blick erkennbar ist.
Kann man fehlende GLP practices am Ende des Projekts nachdokumentieren?
Teilweise dokumentieren: ja. Gleichwertig heilen: nein. Audit-Fähigkeit entsteht während der Studie. Ein starker Abschlussbericht kann Lücken in Rohdatenlogik, Verantwortlichkeiten oder Abweichungen nicht regulatorisch schließen.
Was prüft ein Auditor zuerst?
In der Praxis beginnen Audits auf der Systemebene: Studienplan vorhanden und unterzeichnet? Studienleiter namentlich benannt? Dokumentation der Abweichungen vollständig? Trainingsnachweis vorhanden? Erst danach folgt die inhaltliche Prüfung der Daten. Wer die Systemstruktur sauber aufgestellt hat, hat die härtesten Fragen oft bereits beantwortet.
Müssen Tierstudien für Medizinprodukte immer GLP sein?
Nicht jede Tierstudie muss automatisch vollständig unter GLP laufen. Für frühe Phasen kann GLP-like sinnvoll sein. Bei zentralen Sicherheitsnachweisen steigen die Anforderungen jedoch deutlich. Hier empfiehlt sich eine frühzeitige regulatorische Einschätzung.
Können GLP-Inspektionen auch GLP-like-Studien betreffen?
Formelle GLP-Inspektionen beziehen sich auf registrierte GLP-Prüfeinrichtungen. Behörden und Benannte Stellen können im Rahmen von Einreichungen und Audits auch GLP-like-Studien kritisch auf Nachvollziehbarkeit, Dokumentation und Datenintegrität prüfen und dabei sehr hohe Anforderungen an die Verwertbarkeit der Evidenz stellen.
Quellen & weiterführende Links
- OECD Principles of Good Laboratory Practice
- FDA – 21 CFR Part 58
- EMA GLP Compliance Overview
- BASG – Gute Laborpraxis
Internal links
- GLP vs. GLP-like: Wann GLP, GLP-like oder Non-GLP sinnvoll ist
- Medical Device Compliance nach EU MDR: GLP als Teil der regulatorischen Nachweiskette
- ISO 13485: Qualitätsmanagement und GLP-Dokumentation im MedTech-Labor
- Präklinische CRO auswählen: GLP-Level vor dem Scoping-Gespräch klären
- ISO 9001 für Medizinprodukte: Prozessdokumentation als Brücke zum QMS

Über die Autorin
Dr. med. vet. Henriette Gissel ist Tierärztin und Tierschutzbeauftragte bei Medizin im Grünen. Sie begleitet präklinische In-vivo-Studien im Bereich der Medizinproduktentwicklung mit Fokus auf wissenschaftlich geeignete Tiermodelle, Tierwohl und regulatorisch verwertbare Studiendesigns. Im Rahmen ihrer Tätigkeit unterstützt sie MedTech-Unternehmen bei der Planung, fachlichen Einordnung und Durchführung präklinischer Studien sowie bei der Bewertung möglicher Alternativmethoden im Sinne der 3R-Prinzipien. Ihr Schwerpunkt liegt auf der Verbindung wissenschaftlicher Fragestellungen mit praktikablen und nachvollziehbaren präklinischen Entwicklungsstrategien.
Fachgebiet: Tiermodelle & Studiendesign · 3R-Prinzipien & Alternativmethoden · Präklinische Medizinproduktentwicklung · Präklinische Evidenzstrategien
Stand: April 2026 | Zuletzt geprüft: April 2026