
The most common reason for follow-up requests during regulatory submission: the data package does not withstand regulatory review. Whether 510(k), PMA (Premarket Approval), or MDR conformity assessment (Medical Device Regulation), reviewers assess not only whether preclinical data are available, but whether they are complete, traceable, and integral.
Preclinical studies documented without integrating traceability and data integrity from the outset risks delays, follow-up requests, and the loss of the entire study. Preclinical data must, as far as required for the product, risk, and stage of development, provide a scientifically and regulatory sound basis for the planned clinical investigation.
The FDA has recently underscored the relevance of this topic and, in 2025, established a dedicated webpage for notifications regarding data integrity violations in the medical device sector. This reinforces the stricter requirements for preclinical data.
“Preclinical Data Package” vs. “Nice-to-Have”: What Reviewers Really Expect
What Authorities Understand by a “Preclinical Data Package” (MDR, FDA)
In the regulatory context, preclinical documentation typically includes study protocols, raw data, methodology descriptions, result summaries, and the final report. Under the MDR, where applicable, compliance with Directive 2004/10/EC must be demonstrated.
In practice, this means that every document in the data package must support a clearly defined regulatory rationale. Which components are required depends on the product and the regulatory evidence strategy. Any omitted additional testing must be scientifically justified and traceable.
| Expectation of the Reviewing Specialist | Reality in Many Project Teams | Regulatory Risk |
|---|---|---|
| Complete traceability from protocol to raw data point | Fragmented documentation across different specialist departments | Impossibility of data verification |
| Strict adherence to ALCOA principles | Missing timestamps, handwritten corrections without initials | Potential concerns regarding data integrity |
| Scientific assessment of deviations | Mere listing of errors without analysis of regulatory impact | Doubts regarding study quality and reproducibility |
| Consistent characterization of product, test device, etc. | Minor design changes during the ongoing study | Limited regulatory usability of the study, potentially requiring additional or repeated testing |
ALCOA = Attributable, Legible, Contemporaneous, Original, Accurate
The Operational Reality and Why It Often Falls Short
Data integrity issues remain a recurring focus of regulatory actions. In the medical device sector, the FDA explicitly emphasized this again in 2025. Development teams often provide data packages that appear appear technically sound but fail under structured regulatory review.
Typical gaps include:
- measurement results without reference to the study plan,
- analytical data without documented methodology,
- deviations without assessment of regulatory impact.
Why Data Packages Are Incomplete
Missing Traceability: From Study Plan to Raw Data to Report
The most common systemic cause of incomplete preclinical data is missing traceability. In many preclinical studies, there is no continuous link between the study protocol, collected measurement values, and final report.
In practice, a traceability matrix is one of the most effective tools for systematically demonstrating the required traceability between study plan, raw data, and final report. Without it, reconstructing a study from the final report becomes nearly impossible — and this is often a primary focus during regulatory review.
Unstructured Raw Data Summary Without the ALCOA Principle
Raw data constitute the foundation of every preclinical data package. The ALCOA principle defines the minimum requirements for data integrity: attributable, legible, contemporaneously recorded, preserved in their original form, and accurate.
In practice, many teams struggle with fundamental data integrity requirements: handwritten protocols without dates, overwritten Excel values without audit trails, results without clear attribution to the responsible operator or device. OECD Advisory Document No. 22 (2021) (Organisation for Economic Co-operation and Development) specifies expectations for data integrity and audit trails for GLP-relevant activities (Good Laboratory Practice), especially in electronic systems. ALCOA therefore becomes a review-relevant standard for every preclinical study.
Deviations and CAPAs Not Documented or Not Closed
Deviations from the study protocol are not unusual in preclinical studies. They become problematic only when they are not documented or are documented too late. Under 21 CFR Part 58, deviations from SOPs (Standard Operating Procedures) must be authorized and documented. Protocol changes must be documented, dated, and signed by the study director.
Open CAPAs (Corrective and Preventive Actions) at the time of regulatory submission signal unresolved systemic issues to regulatory reviewers. FDA FY 2022 GLP inspection data show that 45 percent of nonclinical inspections resulted in significant findings. By far the most frequent observation category was failure to follow written procedures. Examples include failure to adhere to approved SOPs, inconsistent process execution, and missing or delayed documentation of required activities.
What a Robust Data Package Must Include at a Minimum
Traceability Process: Study Plan → Raw Data → Analysis → Final Report
A robust preclinical data package is based on a continuous traceability process: study plan (version-controlled and approved before study start) → raw data (ALCOA-compliant and archived without alteration) → statistical analysis (documented analytical methods) → final report (reviewed by quality assurance).
Every step in this chain must reference the previous one. Only this continuous traceability enables complete reconstruction of the study and represents a core prerequisite for GLP compliance and regulatory acceptance.
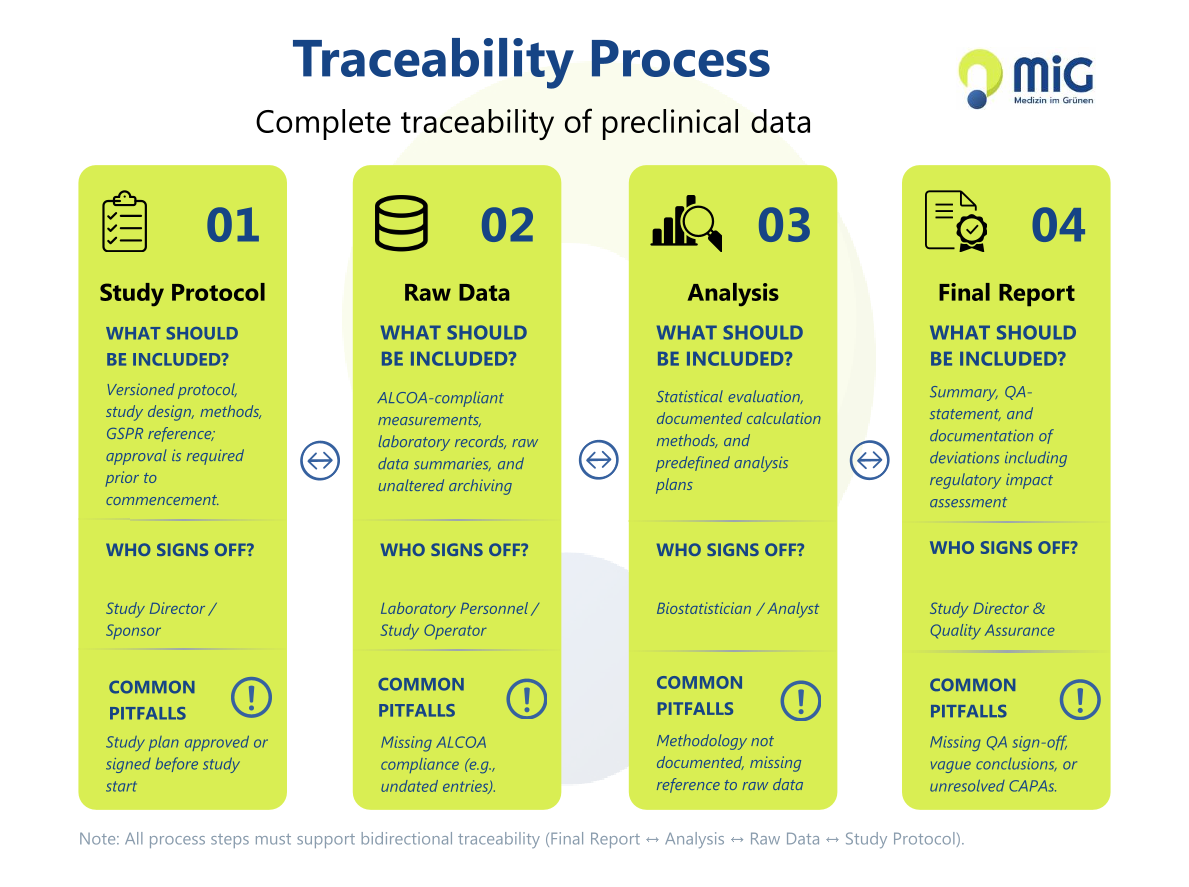
Graphic: “Traceability Process for Preclinical Data”
Minimum Scope of Tables and Appendices
A regulatorily robust preclinical data package typically includes:
- the version-controlled study plan,
- complete and referenced raw data,
- documented analysis plans and statistical evaluations,
- documentation of deviations, including assessment of regulatory impact,
- closed CAPA evidence,
- the final report with quality assurance statement, and
- archiving evidence.
For medical device submissions under the MDR, the connection to the GSPR checklist (General Safety and Performance Requirements) is also required, meaning that preclinical data must explicitly demonstrate which General Safety and Performance Requirements (GSPRs) are addressed.
Overview of the minimum scope of documents and common gaps:
| Document | Common Gap |
|---|---|
| Study plan/version-controlled protocol | Not approved before study start |
| Raw data, complete and referenced | Not ALCOA-compliant, incomplete |
| Analysis plans / statistics | Methodology not documented |
| Documentation of deviations with assessment of regulatory impact | Documented retrospectively during report preparation |
| CAPA evidence, closed | Open CAPAs at regulatory submission |
| Final report with quality assurance statement | Missing quality assurance signature, vague conclusions |
| Archiving evidence | Missing or incomplete |
Summary of Deviations and CAPA Status
Every final report should include a consolidated summary of deviations: When did the deviation occur, what was the cause, what impact did it have on data integrity and regulatory acceptability, and which corrective actions were initiated?
CAPAs should ideally be closed before submission; any remaining actions must be transparently justified, scheduled, and assessed regarding their influence on the usability of the data. This transparency signals methodological maturity and is often expected by reviewers when assessing preclinical data.
How to Identify and Avoid Typical Gaps in Preclinical Data Early
The most effective lever against follow-up requests is the early integration of data integrity into study planning. Teams that implement traceability, ALCOA principles, and deviation management not only when writing the report, but from the first day of the study, avoid typical escalation patterns: retrospective reconstruction of data flows, delayed documentation of deviations, and CAPA follow-up work under time pressure.
In preclinical research, documentation quality must be established from the outset; it is established as a foundation during study planning.
An external review of the data package before regulatory submission systematically identifies remaining gaps and significantly reduces the risk of follow-up requests. This article provides general informational guidance. A project-specific assessment always requires an individual review of the specific data package within the respective regulatory context.
Data Package Review
Is your preclinical data package already ready for submission?
In a structured 30-minute analysis, we assess whether your dossier meets regulatory requirements and specifically identify remaining evidence and documentation deficiencies.
Frequently Asked Questions About Preclinical Data
What is ALCOA in the GLP context?
ALCOA stands for Attributable, Legible, Contemporaneous, Original, Accurate. It describes the minimum requirements for data integrity in GLP-regulated studies. The extended version ALCOA+ adds the criteria Complete, Consistent, Enduring, and Available. OECD Advisory Document No. 22 (2021) further specifies these principles for GLP-regulated activities.
Which raw data do I need to archive?
According to the OECD GLP definition, raw data include all laboratory records and documentation necessary for the reconstruction and evaluation of the study report. This includes handwritten records, electronic measurement data, printouts from measurement devices, photographic documentation, and device device data transferred directly through electronic interfaces. Archiving must ensure long-term readability and availability.
What happens if my data package is rejected during regulatory review?
If data packages are incomplete or deficient, authorities may issue formal follow-up requests, such as a request for additional information by the FDA or a nonconformity letter from the Notified Body. In the most serious case, for example where data integrity violations are proven, the FDA may reject all study data from a laboratory.
Does ALCOA apply to medical devices or only to pharmaceuticals?
ALCOA applies to all GxP-regulated areas (GMP, GLP, GCP, GDP), including preclinical testing of medical devices. EU MDR Annex II, Section 6 explicitly refers to GLP Directive 2004/10/EC. The FDA also applies 21 CFR Part 58 (GLP) to nonclinical safety studies for medical devices. These principles are therefore equally applicable in the medical technology sector as they are for pharmaceutical developers.
Sources & Further Reading
External References
- OECD – Advisory Document No. 22: GLP Data Integrity (2021)
- FDA – Notifications on Data Integrity – Medical Devices (2025)
- FDA – Recommended Content and Format of Non-Clinical Bench Performance Testing Information in Premarket Submissions (2019)
- EU MDR 2017/745
- EU GLP Directive 2004/10/EC
Internal Links
- Preclinical Evidence: EU MDR, FDA, and regulatory roadmap
- Medical Device Regulation: How preclinical data fits into the MDR architecture
- Technical Documentation for Medical Devices: The data package as part of the Technical File
- GLP-compliant practices: GLP as a quality marker for robust data packages
- Anatomical Analysis: Histological findings as a critical part of the data package
- Medical Device Commercialization: Robust data packages as a time-to-market accelerator

About the author
Dr. med. vet. Henriette Gissel is a veterinarian and Animal Welfare Officer at Medizin im Grünen. She supports preclinical in vivo studies in medical device development with a focus on scientifically appropriate animal models, animal welfare, and regulatory-relevant study design. In her role, she works with MedTech companies on the planning, scientific evaluation, and conduct of preclinical studies, as well as on the assessment of potential alternative methods in line with the 3R principles. Her work focuses on aligning scientific objectives with practical and scientifically sound preclinical development strategies.
Areas of expertise: Animal Models & Study Design · 3R Principles & Alternative Methods · Preclinical Medical Device Development · Preclinical Evidence Strategies
Status: May 2026 | Last reviewed: May 2026